

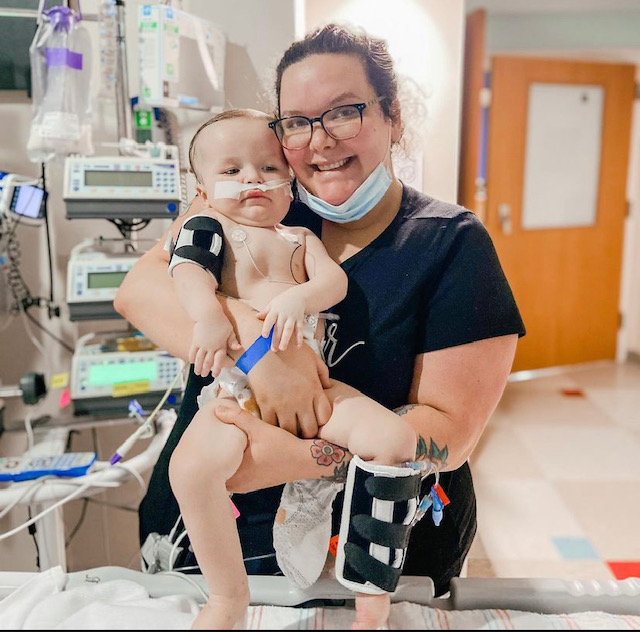
 As I read “the average life expectancy of an infant diagnosed with type 1 SMA is less than 2 years old,” I felt my stomach turn. “Don’t google it,” the neurologist told me. But I didn’t even make it home before doing so. This couldn’t be real. We were still living in the high of having a newborn. Our first baby. There was no way that something so terrible could have been there all along. But it would only take a few short days and an expedited genetic test to confirm our worst fear. Blaise indeed did have Spinal Muscular Atrophy, Type 1 (also known as Werdnig-Hoffman disease). We were in for a wild next two years with a lot of lows, but what wasn’t expected were all the beautiful and unbelievable highs to come with it.
As I read “the average life expectancy of an infant diagnosed with type 1 SMA is less than 2 years old,” I felt my stomach turn. “Don’t google it,” the neurologist told me. But I didn’t even make it home before doing so. This couldn’t be real. We were still living in the high of having a newborn. Our first baby. There was no way that something so terrible could have been there all along. But it would only take a few short days and an expedited genetic test to confirm our worst fear. Blaise indeed did have Spinal Muscular Atrophy, Type 1 (also known as Werdnig-Hoffman disease). We were in for a wild next two years with a lot of lows, but what wasn’t expected were all the beautiful and unbelievable highs to come with it.
And that’s what brings us to today. As I am sitting here writing this, Blaise is working through his second therapy of the day. This time it’s occupational therapy where he and Ms. Karen (who has been with him since he was 10 months old) are SITTING together and coloring. Yes, sitting… independently I might add. Something no individual with type 1 SMA was ever supposed to be able to do. And how old is Blaise now? He is 2 years & 4 months old and the picture of health. Another miraculous goal he was not supposed to reach.
So what changed? Did google lie? Surely not. Well, you see what’s changed is SMA and what the quality of life looks like for those diagnosed. In December of 2016, the first treatment for SMA was FDA approved for patients of all ages. This drug was the beginning of a huge movement in advances in modern medicine that got us to where we are today. Not too long after that, a groundbreaking gene therapy was FDA approved for children under the age of two diagnosed with SMA, and this drug is exactly what Blaise’s team had their eyes set on even before we got the official diagnosis. We KNEW this was the best chance to stop this progressive neuromuscular disease dead in its tracks. And we needed to move fast because every day that went by Blaise was getting weaker. He was losing more movement, showing signs of respiratory distress and unknown to us at the time, he was losing his ability to swallow. So just two weeks after his diagnosis, 14-week-old Blaise was in my arms with two IVs in his head, running the world’s most expensive drug through his tiny little veins. Saving his life.
I wish I could tell you that day was it, that SMA was behind us after that. But this was a treatment, not a cure. Let’s be clear, there still isn’t one. So the next two years of his life were anything but easy. Unfortunately, back in 2020, Texas didn’t test for SMA on their Newborn Screening (as of June 2021 they now do), so by the time we were diagnosed, Blaise was VERY symptomatic. We had our work cut out for us in hopes to regain a lot of the muscle lost in those first 4 months of life. Immediately we started him in Physical, Occupational and Speech Therapy. We went inpatient to our Children’s hospital about a month after treatment to get all the necessary medical equipment we now needed at home for our day-to-day life. We also discovered at that time that Blaise indeed had lost his ability to swallow safely and therefore stopped feeding him orally and placed a g-tube (gastrostomy tube). The following year he had several hospital stays, the worst one being 6 weeks long with multiple intubations from a simple cold. His muscles used to breathe just weren’t strong enough to support himself during the illness. But even with all this chaos, we went to bed every single night clinging to the relief that just 4-5 years earlier, we would have been told to take our baby home and love him until his body failed him.
Then came August of 2020. As if two treatment options for SMA wasn’t amazing enough, a THIRD treatment was now FDA approved, and this one we had been eagerly waiting for. After diagnosis, I did what a lot of parents would do and completely submerged myself into the SMA community and research. Upon doing so, I started reading up on a drug called Risdiplam (now known as Evrysdi) that was in clinical trials, and I just knew Blaise had to have it. Without going super nerdy on you, it was the missing puzzle piece. The gene therapy had gone in and essentially replaced Blaise’s missing/mutated gene, but he still created very little SMN protein crucial to make gains. So in comes Evrysdi, helping increase and maintain those SMN protein levels. The best part? This drug was a liquid form prescription delivered to our door and administered daily via g-tube. No pokes, no IVs, no doctor’s office visits. Isn’t modern medicine cool? So, with the help of Blaise’s very supportive neurologist, we were able to start him on Evrysdi in October of 2020. And let me tell you, the changes were apparent in the first week. His energy and stamina doubled. He began to have more movement in his limbs, especially his legs. And we give a lot of credit to Evrysdi for his massive leap in oral strength. His speech is on track as any average 2-year-old. I mean the kid literally doesn’t stop talking.
I cannot fathom what an SMA diagnosis felt like 5 years ago. And I try not to let myself go there. But when I do, I tear up thinking of the wonderful families that have come before mine that put their kids or self through these clinical trials with drugs they weren’t sure would work, in hopes to find any help for their loved ones or those to come after them. And I think of the families that came before the clinical trials that advocated, even while grieving the loss of their child, for research to find these treatments. Because those are the real heroes of this story. If it wasn’t for them and some really, really smart scientists and geneticists, I don’t think I’d be sitting here watching my happy 2-year-old roll around on the floor chunking toys across the room. And for that, I am so thankful.



